Blogger: Åsmund Flobak,
stipendiat ved Institutt for kreftforskning og molekylær medisin

Mitt forskningsprosjekt handler om å forutsi gode kombinasjoner av kreftmedisiner. Og for å si dette mer presist: Jeg leter etter synergistiske effekter, hvor to medisiner sammen har en kraftigere effekt enn hva man skulle tro ved bare å legge sammen effektene fra hver medisin. Slike kombinasjoner er ettertraktede fordi de vil nemlig tillate høyere effekt mot f.eks vekst av kreftceller enn dosen (av hver medisin) skulle tilsi. Bivirkninger er ofte avhengige av dose, så hvis dose (og bivirkning) kan reduseres selv om effekten økes, vil det være gunstig.

Åsmund Flobak på laben. (Foto: Antonsen/Adresseavisen)
I artikkelen som nettopp kom ut i tidsskriftet PLoS Computational Biology har vi funnet en metode for å finne synergier. Metoden er basert en datamodell som kan simulere kombinasjonsmedisiner. Vi bruker metoden på 21 kombinasjoner av kreftmedisiner, og forutsier at fem av disse er synergistiske – for fire av de fem stemmer det. To av disse synergiene er velkjent og prøves i dag ut i mange kliniske studier, mens de to siste synergiene ble publisert for første gang med vår forskningsartikkel.
De medisinene vi jobber med er medisiner som påvirker signaltrafikken i en celle, dvs de signalene som sammen styrer avgjørelser cellene hele tiden tar, som “skal jeg vokse nå?”. Disse beskjedene sendes og tolkes ved hjelp av signalstoffer, som proteiner og rna. Denne typen medisiner skiller seg fra cellegift, og noen slike medisiner brukes allerede på sykehusavdelingene – og det er forventet de vil bli stadig viktigere i behandling av kreft (og andre sykdommer som f.eks immunologiske/revmatiske sykdommer).
Noen kombinasjoner av ulike kreftmedisiner gir ekstra god effekt. (illustrasjonsbilde: iStockPhoto).
For kreftcellene er puslespillbitene molekyler, gener, mRNA og proteiner. Vi har i dag muligheten til å analysere veldig mange slike biter – og en utfordring i dag blir at vi kan måle så mange biter, at ingen klarer å sette sammen hele puslespillet. Her kommer datamodeller av kreftceller inn, for hvis en datamaskin bare vet hvordan den skal se på en bit så kan den også hjelpe oss å legge sammen puslespillet. Men hva skal vi se etter når vi vil se hvordan to biter passer sammen?
Forskningsprosjektet er et samarbeid mellom Institutt for biologi (biveileder Martin Kuiper) og Institutt for Kreftforskning og molekylær medisin (hovedveileder Astrid Lægreid). Sammen med andre forskere ved NTNU, i Paris og i Marseille, har jeg laget en datamodell over signalveier i en celle. Vi har brukt boolsk matematikk (også kalt diskret matematikk) som grunnlag for signalreguleringen. Boolsk matematikk er ja/nei-matte, noe er enten «mer» eller «mindre», «av» eller «på». Denne matematikken passer perfekt til dagens forståelse av biologi, som foreløpig er preget av kvalitative analyser og ikke kvantitative.
Først laget jeg et generelt signaloverføringskart over kjente signalveier i en celle. Jeg studerer kreftceller fra magesekk, og har notert hvilke signalproteiner som er aktive i de cellene jeg studerer. Så oppdaterte jeg signalreguleringen i modellen til den stemmer med molekylære tilstander målt i magekreftceller. Datasimuleringene er altså basert på “biomarkører” målt i cellelinjene, dvs at vi i prinsippet har et signaloverføringskart som er spesialtilpasset kreftcellene vi studerer.
Jeg har sett på effekten av å blokkere parvis to og to signalproteiner i datamodellen, og med datamodellanalysene identifiserte jeg fem potensielt overlegne kombinasjoner blant 21 kombinasjoner totalt. Jeg var rimelig spent da jeg testet min første foreslåtte kombinasjon på kreftceller i desember 2013 – og da jeg hadde bekreftet én slik kombinasjon var det et perfekt tidspunkt for juleferie – det er mitt største øyeblikk på laboratoriet og jeg kunne nesten ikke tro det jeg så – men jeg fant det samme igjen og igjen når jeg repeterte eksperimentet.
Det viste seg at fire av fem kombinasjoner predikert av modellen var overlegne også i praksis, det var litt av en følelse å se hvordan all fiklingen, tenkingen og kaffen på kontoret ble omgjort til noe som faktisk funket. Og vel så viktig, alle de resterende kombinasjonene som vår databaserte analyse predikerte å være dårlige viste seg å være dårlige også i praksis.
Det viste seg så at andre forskere hadde funnet to av disse kombinasjonene før meg, og de to kombinasjonene er det stort fokus på i dag – de prøves ut i over 20 kliniske studier. De to siste kombinasjonene er det enda ingen andre som oppdaget – vi var først ute.
Det jeg er mest fornøyd med er metoden jeg har vært med å utvikle, hvor vi kombinerer datamodellanalyser med eksperimentelle data (biomarkører), for å finne nye gode medikamentkombinasjoner for andre kreftceller.
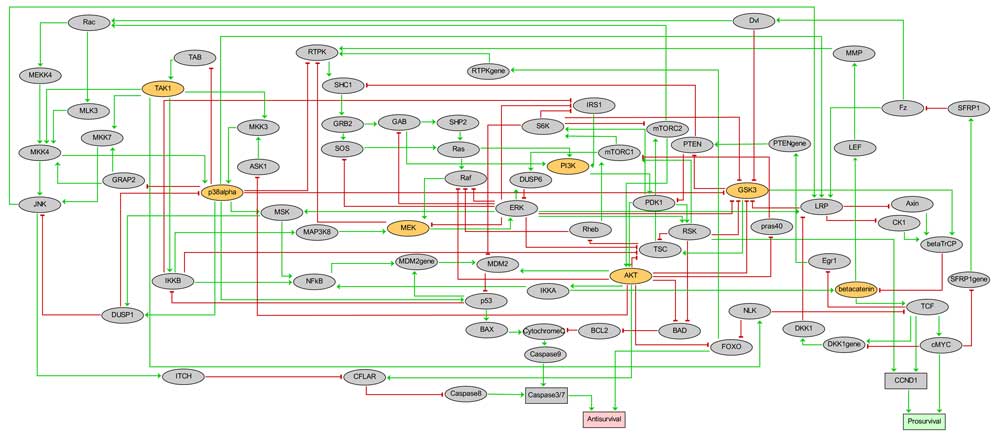
Figuren viser en full oversikt over signaltrafikkartet vi har brukt, der de grå boksene er gener/proteiner, og de gule boksene er de proteinene vi har slått ut med signalhemmere. Aktivitetssignaler i hvert protein vil overføres via grønne og røde koblinger, der de grønne gir aktiverende signaler og de røde gir hemmende signaler. Signalene samles til slutt i de to fargede firkantede boksene nederst, hvor den røde integrerer veksthemmende signaler og den grønne integrerer vekstfremmende signaler. Datamodellen hjelper oss å holde totaloversikten over signalene, og brukes videre til å simulerer medikamenter i kombinasjoner, hvor vi i våre forsøk parvis har slått ut de gule proteinene.
Portrettfoto: Sivelin Kjølstad